GLMM and Hierarchical Bayesian Models
Partial pooling for stable estimation of hospital-level SSI risk
Random-intercepts GLMM¶
Facilities are nested within counties, creating a hierarchical data structure in which outcomes from facilities in the same county may be correlated due to shared local factors such as patient populations, referral patterns, or reporting practices.
To account for this clustering, a binomial generalized linear mixed model (GLMM) with a random intercept for county and fixed effects for facility type can be fit to the data. This specification allows the baseline log-odds of infection to vary across counties while estimating the average effect of facility type across all counties.
Each county is assigned a random intercept that represents its deviation from the overall mean log-odds. The model can be written as:
where
is the probability of SSI for facility in county
is the overall intercept
is the fixed effect of facility type
is the county-level random intercept
The following R code fits this GLMM to the observed data and displays the results.
Source
# Random intercepts glmm
glmm_fit <-
glmer(cbind(Infections_Reported, No_Infections) ~ Facility_Type +
(1 | County), data = colon_fac_ach, family = binomial)
summary(glmm_fit)Generalized linear mixed model fit by maximum likelihood (Laplace
Approximation) [glmerMod]
Family: binomial ( logit )
Formula: cbind(Infections_Reported, No_Infections) ~ Facility_Type + (1 |
County)
Data: colon_fac_ach
AIC BIC logLik -2*log(L) df.resid
943.3 961.6 -466.6 933.3 283
Scaled residuals:
Min 1Q Median 3Q Max
-2.0170 -0.7076 -0.3761 0.3658 6.6185
Random effects:
Groups Name Variance Std.Dev.
County (Intercept) 0.1779 0.4218
Number of obs: 288, groups: County, 42
Fixed effects:
Estimate Std. Error z value Pr(>|z|)
(Intercept) -4.4636 0.2175 -20.523 < 2e-16 ***
Facility_TypeCommunity, >250 Beds 0.6328 0.2283 2.772 0.00556 **
Facility_TypeCommunity, 125-250 Beds 0.3296 0.2373 1.389 0.16478
Facility_TypeMajor Teaching 0.6146 0.2152 2.855 0.00430 **
---
Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1
Correlation of Fixed Effects:
(Intr) F_TC>B F_TC1B
Fc_TC,>250B -0.834
F_TC,125-2B -0.786 0.783
Fclty_TypMT -0.882 0.887 0.829The random-intercepts GLMM yields results that are broadly consistent with those from the standard logistic regression and Bayesian binomial models. Facilities classified as community hospitals with more than 250 beds and major teaching hospitals exhibit significantly higher odds of surgical site infection relative to the reference group, while the effect for community hospitals with 125 to 250 beds is positive but not statistically significant.
The estimated county-level random intercept variance is 0.178, corresponding to a standard deviation of 0.422 on the log-odds scale, indicating meaningful heterogeneity in baseline infection risk across counties. This suggests that unobserved county-level factors contribute to variation in infection rates beyond what is explained by facility type alone.
Comparing AIC values with the logistic model provides a rough measure of relative model fit. Although the models differ in structure and are estimated using different likelihood formulations, the large difference observed here, 978 for the facility-type-only logistic regression versus 943 for the GLMM, indicates that the model including a county-level random intercept fits the data substantially better.
Partial pooling¶
The estimated in the GLMM are empirical Bayes estimates of the random intercepts, commonly referred to as BLUPs. Each BLUP is a weighted combination of the observed county mean and the overall mean, implementing partial pooling: counties with few facilities are shrunk strongly toward the global mean, while counties with many facilities rely more on their own data. Formally, for a simplified normal approximation, the BLUP can be expressed as:
where
is the estimated random intercept for county (BLUP)
is the observed mean for county
is the overall mean
is the number of facilities in county (more facilities → less shrinkage)
is the residual variance at the facility level
This weighting shows that small counties are pulled toward the overall mean, while larger counties contribute more of their own information. The GLMM thus provides conditional estimates with partial pooling, stabilizing predictions for low-volume counties.
Bayesian hierarchical models implement the same partial pooling concept. They additionally produce full posterior distributions for each parameter, allowing richer probabilistic interpretation.
Hierarchical Bayesian binomial model¶
The hierarchical Bayesian binomial model is specified by the following set of probability distributions and link function.
The regression coefficients and the county-level mean are assigned weakly informative normal priors with mean 0 and standard deviation 2.5. The standard deviation of the county-level effects has a positive-truncated Student- prior with 3 degrees of freedom and scale 2.5. These choices follow Gelman et al. (Bayesian Data Analysis, Section 5.7) and are intended to provide stability for the hierarchical variance while allowing the data to dominate the posterior Gelman et al., 2013.
JAGS model syntax¶
Below is the JAGS syntax for fitting this model in R.
Source
## Hierarchical Bayesian binomial model
# Transform data for JAGS; use Community, <125 Beds for reference level
data_jags_h <- list(
y = colon_fac_ach$Infections_Reported,
n = colon_fac_ach$Procedure_Count,
county = colon_fac_ach$County_numfactor,
beds_125to250 =
as.numeric(colon_fac_ach$Facility_Type == 'Community, 125-250 Beds'),
beds_gt250 =
as.numeric(colon_fac_ach$Facility_Type == 'Community, >250 Beds'),
major_teach = as.numeric(colon_fac_ach$Facility_Type == 'Major Teaching'),
N = nrow(colon_fac_ach),
J = length(unique(colon_fac_ach$County)), # J = 42 counties
K = 3 # number of predictors
)
# JAGS syntax
mod_string_h <- " model {
# likelihood
for (i in 1:N) {
y[i] ~ dbin(p[i], n[i])
logit(p[i]) <- a[county[i]] + b[1]*beds_125to250[i] + b[2]*beds_gt250[i] +
b[3]*major_teach[i]
}
# random intercepts for counties (implemented with prec_a = 1 / sigma^2)
for (j in 1:J) {
a[j] ~ dnorm(mu, prec_a)
}
# priors for betas
for (k in 1:K) {
b[k] ~ dnorm(0.0, 1.0/2.5^2) # weakly informative prior (sd ~2.5)
}
# hyperpriors for random intercepts
mu ~ dnorm(0.0, 1.0/2.5^2)
# per Gelman's Bayesian Data Analysis section 5.7:
sigma ~ dt(0, 1/2.5^2, 3) T(0,)
prec_a <- 1 / sigma^2
} "
# Model setup
set.seed(456)
# Define seeds for JAGS chains
jags_seeds <- list(
list(.RNG.name = "base::Wichmann-Hill", .RNG.seed = 457),
list(.RNG.name = "base::Wichmann-Hill", .RNG.seed = 458),
list(.RNG.name = "base::Wichmann-Hill", .RNG.seed = 459)
)
mod_h <- jags.model(textConnection(mod_string_h),
data = data_jags_h,
inits = jags_seeds,
n.chains = 3)
update(mod_h, 1e4) # burn-in
params_h <- c("b", "mu", "sigma", "a")
# Run model
mod_sim_h <- coda.samples(mod_h, variable.names = params_h, n.iter = 1e5)Compiling model graph
Resolving undeclared variables
Allocating nodes
Graph information:
Observed stochastic nodes: 288
Unobserved stochastic nodes: 47
Total graph size: 1997
Initializing model
Gelman-Rubin¶
For brevity, only the Gelman–Rubin convergence diagnostics are shown for the main parameters.
All parameters including the 42 random effects have potential scale reduction factors close to 1, with a maximum value of 1.01. This indicates good mixing and no evidence of lack of convergence across chains for either the county-level random effects or the hyperparameters. The posterior samples are therefore stable for inference.
Deviance information criterion (DIC)¶
The DIC statistic for the model is shown below.
Mean deviance: 899.4
penalty 24.55
Penalized deviance: 924 The penalty is much smaller than the actual number of parameters because the county effects are not estimated independently. Instead, they borrow strength from one another through the hierarchical structure, which effectively reduces the model’s complexity.
For comparison, the penalized DIC for the non-hierarchical Bayesian model was 978.3 (see non-hierarchical model DIC). This lower value for the hierarchical model indicates a substantially better fit to the data after accounting for model complexity.
Residuals plot¶
Pearson residuals for the hierarchical Bayesian binomial model plotted against fitted probabilities are shown below.
Figure 4. Pearson residuals vs fitted probabilities
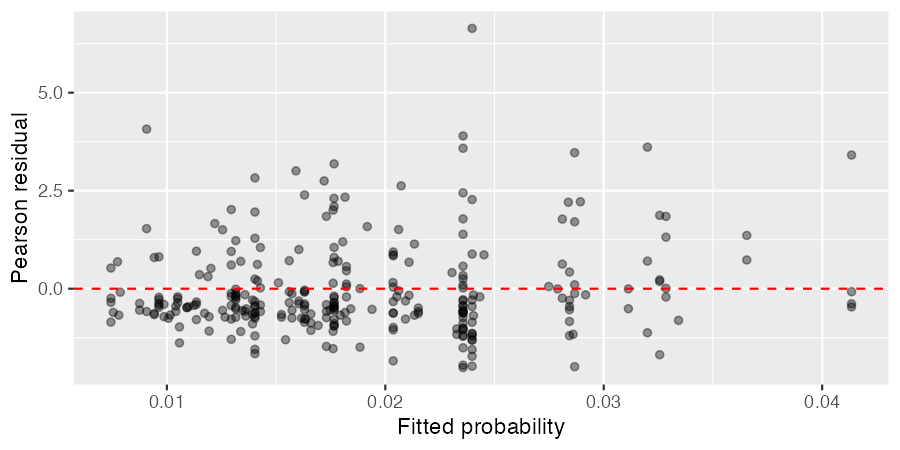
Most residuals are close to zero with no obvious trend, indicating the model captures the overall structure of the data well. One facility shows a relatively large residual and is more influential than the others, while the remaining residuals are spread along the fitted values as expected given the hierarchical partial pooling.
Model summary¶
The results of the fitted model are shown below. For brevity, the county random effects are not shown.
Source
# Show only fixed effects and hyperparameters (exclude random effects)
params_to_show <- c("b[1]", "b[2]", "b[3]", "mu", "sigma")
summary(mod_sim_h[, params_to_show])
Iterations = 11001:111000
Thinning interval = 1
Number of chains = 3
Sample size per chain = 1e+05
1. Empirical mean and standard deviation for each variable,
plus standard error of the mean:
Mean SD Naive SE Time-series SE
b[1] 0.2989 0.2348 0.0004287 0.0036989
b[2] 0.6113 0.2254 0.0004115 0.0038306
b[3] 0.5934 0.2125 0.0003880 0.0038373
mu -4.4482 0.2171 0.0003965 0.0036734
sigma 0.4633 0.1023 0.0001868 0.0006561
2. Quantiles for each variable:
2.5% 25% 50% 75% 97.5%
b[1] -0.1530 0.1396 0.2956 0.4551 0.7683
b[2] 0.1822 0.4574 0.6076 0.7601 1.0669
b[3] 0.1898 0.4481 0.5894 0.7336 1.0252
mu -4.8906 -4.5907 -4.4429 -4.2995 -4.0391
sigma 0.2912 0.3911 0.4533 0.5244 0.6922
Compared with the reference group (Community, <125 Beds), all three facility types have higher estimated probabilities of infection. On the log-odds scale, the posterior means are 0.30, 0.61, and 0.59 for Community 125–250 Beds, Community >250 Beds, and Major Teaching facilities, respectively. The 95% credible intervals for all three groups lie mostly above zero, suggesting that each facility type is likely associated with higher infection probability than the reference.
The overall county-level intercept has a posterior mean of –4.45, reflecting the baseline log-odds for the reference facility type, while the posterior standard deviation of the county-level random effects, 0.46, indicates moderate variation in baseline risk across counties.
Summary¶
The hierarchical Bayesian model provides partially pooled estimates of SSI risk for each facility while accounting for county-level variation. The posterior means indicate that all three non-reference facility types have higher log-odds of infection than the reference group, with 95% credible intervals largely above zero. The county-level standard deviation, σ ≈ 0.46, reflects moderate variation in baseline risk across counties. Compared with simpler models, the hierarchical approach stabilizes estimates for smaller facilities and better captures multilevel structure, offering a principled framework for facility-level risk estimation.
These results provide an initial view of facility- and county-level effects; a more formal comparison across models, along with further diagnostics and residual analysis, can be conducted in a future review. For now, this establishes a foundation for interpreting partial pooling and posterior uncertainty in SSI risk estimates.
- Gelman, A., Carlin, J. B., Stern, H. S., Dunson, D. B., Vehtari, A., & Rubin, D. B. (2013). Bayesian Data Analysis (3rd ed.). Chapman. 10.1201/b16018